このページは基本的にはVASP 4.6の経験に基いて書かれているため,古い情報かもしれないので注意.
VASPとは,平面波基底を用いた密度汎関数理論(Density Functional Theory)計算を行う有償プログラムである.
vasp.4.lib
まずはvasp.4.libを最初にコンパイルしなければいけないらしい.
$ cd vasp.4.lib
$ cp makefile.????_???? makefile
$ make
とすることで,コンパイルが始まり,libdmy.aというライブラリが作られる.
vasp.4.6
次に,vasp.4.6ディレクトリに移り,makefileを作る.
$ cp makefile.????_??? makefile
makefile内をいろいろと変更する必要があるらしい.
serial executable
シリアル計算用のコンパイルのためには,
FC= ifort
BLAS=-L/opt/intel/mkl/10.2.6.038/lib/em64t -lguide -lpthread
FFT3D= fftw3d.o fft3dlib.o /opt/fftw-3.3.2/lib/libfftw3.a
のように,IntelのMKLライブラリを指定する.lib/32とlib/64とlib/em64tのどれを使うべきなのかは定かではないが,ココではem64tでないと,
ld: skipping incompatible: ....
のようなエラーが出てコンパイルできなかった. また,lib/em64tの中で,libmkl_blas95.a,libmkl_intel.a,libmkl_lapack95.aを参照するが,それらが存在しなかったので,
$ ln -s libmkl_blas95_lp64.a libmkl_blas95.a
$ ln -s libmkl_intel_lp64.a libmkl_intel.a
$ ln -s libmkl_lapack95_lp64.a libmkl_lapack95.a
のようにシンボリック・リンクを貼った. また,ここでは,FFTのライブラリとしてFFTWを用いる設定になっている.このとき,コンパイル中に,
include 'fftw3.f'
の fftw3.f が見つからないと言われるかもしれない. そんなときは,
cp /opt/fftw-3.3.2/include/fftw3.f .
としてFFTWによって提供されているソースファイルをコピーして置いておく.
parallel executable
並列計算のためには,
FC= /opt/intel/impi/4.0.0.028/intel64/bin/mpiifort
BLAS=-L/opt/intel/mkl/10.2.6.038/lib/em64t -lguide -lpthread
FFT3D= fftmpiw.o fftmpi_map.o fft3dlib.o /opt/intel/Compiler/11.1/073/mkl/interfaces/fftw3xf/libfftw3xf_intel.a
のように,mpiifortをcompilerに指定し,fft3dとしてmpi用のものを指定.(しかも,MKLのラッパーを用いている.そんな必要はないと思われるが.) さらに,CPPの並列計算用の部分をuncommentする.
CPP = $(CPP_) -DMPI -DHOST=\"LinuxIFC\" -DIFC \
-Dkind8 -DNGZhalf -DCACHE_SIZE=4000 -DPGF90 -Davoidalloc \
-DMPI_BLOCK=500 \
-DRPROMU_DGEMV -DRACCMU_DGEMV
これで
$ make
とすれば,とりあえずコンパイルはできる.
Run vasp
INCAR,POTCAR,POSCAR,KPOINTSのあるディレクトリで,
$ vasp
とすれば実行され,OUTCAR,OSZICAR,IBZKPT,CHGCARなどの出力ファイルがでてくる.
Parameters in INCAR file
User guideに詳しく記述されているのでそちらを参考にすべし.
ENCUT
エネルギーカットオフ.逆格子空間の波数Gの二乗の上限値を決めるもので,精度と計算量(計算時間)に直接影響する.ポテンシャルファイル(POTCAR)内にENMAXおよびENMINとしてデフォルトのカットオフが記載されているので,それを使うことが推奨されている.原理的にはENCUTが大きければそれだけ精度が上がるはず.
EDIFF
初期値:1.0e-4 SCF計算の収束を判定するエネルギー差の値.これが小さければ厳しい収束判定となるので,精度が高くなり,計算時間も増す.
ISMEAR
Fermi levelを決める際に行うsmearingの手法の指定.
- 金属以外ではpartial occupancyが発生しないので,ISMEAR=-5 (tetrahedron method with Blochl correction)が良いらしい.
- 金属の場合は,ISMEAR=1 or ISMEAR=2 で,SIGMA=0.2 (default)が良い? SIGMAの値は経験的?
POTIM
IBRIONの値によって違う意味を持つようだが,IBRION=3(damping MD)を行う場合は,時間刻み(fs)を意味するので,0.5程度が妥当なようだが,水素があるような系では小さくとるべきかもしれない.
Postprocess
Charge density visualization using VESTA
VESTAという可視化プログラムを利用する. CHGCARとOUTCARを同一フォルダに入れておいて,CHGCARファイルをVESTAへドラッグアンドドロップすれば,表示してくれる(MacOSX).
VASPのCHGやCHGCAR,PARCHGなどのデータは単位なし(電子の個数)となっている(つまり計算セルの体積が掛けられている).VESTAではそれを計算セルの体積で割った値を採用している.その際の体積の単位がBohr^3なので,VESTAでの電子密度データの値は1/Bohr^3なので注意.
Density of states
まず,MDやrelaxationなどを行った結果表示されるようなDOSは「usually useless」らしい.
ISTART = 1
NSW = 0
として再計算したDOSCARを用いるべしとのこと. 実際に高精度なDOSを描きたい場合には,KPOINTSを多少増やさないといけないかもしれない.SCF計算の収束値(EDIFF)もrelaxation計算よりは厳しくするべきと思われる. その際,
ICHARG = 11
のように,ICHARGに「+10」することでself-consistent計算をしないように設定できる.(これはすべてのelectronic minimizationの間,charge densityひいてはpotentialを固定しておくということ.)
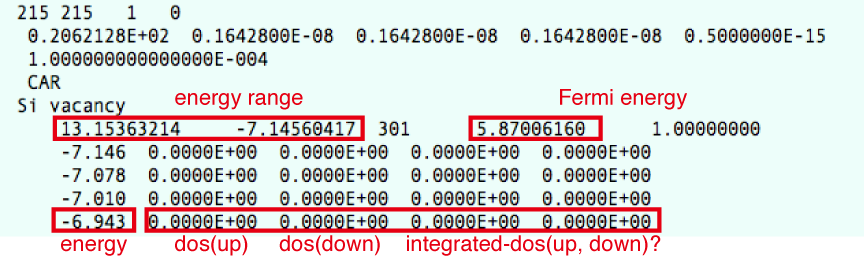
DOSCARというファイルに出力される.spin-polarizedの場合とnon-polarizedの場合で出力が多少異なる.polarizedの場合は,upとdownのDOSが含まれる. spin non-polarized (ISPIN=1)の場合のDOSのリストのところは以下のような記述になっている.
energy DOS integrated-DOS
spin-polalized (ISPIN=2)の場合は,
energy DOS(up) DOS(down) integrated-DOS(up) integrated-DOS(down)
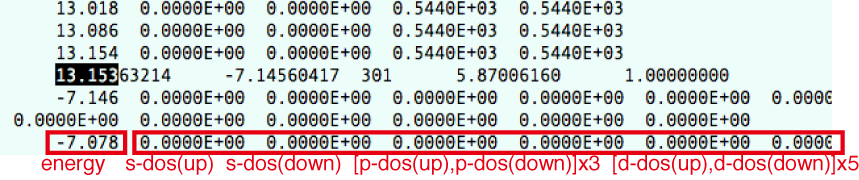
LORBITの値をPDOSを出力するように設定してある場合は,この後に,
energy s-DOS(up) s-DOS(down) p-DOS(up) p-DOS(down) d-DOS(up) d-DOS(down)
のように出力される(spin-polarizedの場合).
実際のDOSCARファイルの中身はこんな感じ.
partial (band decomposed) charge density
すでに計算が行われて収束していること(WAVECARファイルが存在していること)を前提としている.
参考:http://cms.mpi.univie.ac.at/vasp/guide/node145.html
INCARファイルに以下のような行を追加して再計算する.
LPARD = .TRUE.
NBMOD = -2
EINT = 5.67 5.8700616
LSEPB = FALSE
LSEPK = FALSE
LPARD:partial charge densityを計算するというflag.これを立てるとpartial charge densityのみ計算して終了する.NBMOD:どこのpartial charge densityを計算するかという値.- 「-1」はIBANDで指定されたバンドのpartial charge density
- 「-2」はEINTで指定されたenergy range内のpartial charge density