VASPは,平面波基底を用いた密度汎関数理論(Density Functional Theory)計算を行う有償プログラム. 基本的にはRef.[1]のVASP wikiページを参照すること.このページは個人的なメモなのであしからず.
VASP 5.3.5のアカデミックライセンスは,現在(20150116時点)では4000EUROほどするはず.
VASPが同梱されているMedeAというソフトウェアはもっと高く,10倍位の値段だったと思う. DFT計算だけでなく,モデリングから解析までを助けるGUIがセットになっているから高いのかも.
インストール方法は,バージョン毎に別の場所に記載する.
Running VASP
INCAR,POTCAR,POSCAR,KPOINTSのあるディレクトリで,
$ vasp
とすれば実行され,OUTCAR,OSZICAR,IBZKPT,CHGCAR,vasprun.xmlなどの出力ファイルがでてくるはず.
vasprun.xmlには計算条件や結果の情報など多くが含まれており,後処理のスクリプトなどで扱いやすいので,最も重要なファイル.
並列実行したい場合は,
$ mpirun -np 8 vasp
などとすればよい.
INCAR内に並列数に関する変数NCOREやKPARがある.
Parameters in INCAR file
User guideに詳しく記述されているのでそちらを参考にすべし.
ENCUT
エネルギーカットオフ.逆格子空間の波数Gの二乗の上限値を決めるもので,精度と計算量(計算時間)に直接影響する.ポテンシャルファイル(POTCAR)内にENMAXおよびENMINとしてデフォルトのカットオフが記載されているので,それを使うことが推奨されている.原理的にはENCUTが大きければそれだけ精度が上がるはず.
応力の精度はENCUTの値に大きく依存する(VASP workshop資料参照).POTCAR内のENMAXの値から30%程度大きなENCUT値にしないと誤差が大きい場合もある.そのため,セルの最適化を行う際は大きめのENCUTを使う必要がある.セル内の原子座標緩和はもう少し小さい値でもいいらしい.
EDIFF
初期値:1.0e-4
SCF計算の収束を判定するエネルギー差の値.これが小さければ厳しい収束判定となるので,精度が高くなり,計算時間も増す.
一般に,原子にかかる力はエネルギーよりも誤差が大きい傾向があるので,力の精度を求める場合にはより厳しいEDIFFの値(例えば1.0E-6)が必要となる.
ISMEAR
KPOINTSに記載.
SIGMA
軌道に電子を詰める際のFermi順位付近のぼかしの幅(eV単位?).上のISMEARの方法と関係がある.あまり小さな値だとFermi順位近傍の電子の入る軌道と入らない軌道の変化が激しくなりすぎて,SCFの収束が悪くなるらしい.そのため,この値は大きく設定するのが良いらしい.しかし,計算結果のentory T*Sの値が十分小さく(< 1-2 meV/atom)なるよう注意する.
POTIM
IBRIONの値によって違う意味を持つようだが,IBRION=3(damping MD)を行う場合は,時間刻み(fs)を意味するので,0.5程度が妥当なようだが,水素があるような系では小さくとるべきかもしれない.
NCORE, NPAR
並列計算の際の並列化の仕方の指定.
NCORE = (num of cores)/NPAR/KPAR という関係にあり,NCOREは一つの軌道の計算にどの程度のコアを使うかという指定のようで,大体 NCORE = sqrt(num of cores) 程度とするのがいいらしい.
See: NCORE @VASP Manual,
IMIX, AMIX, BMIX, AMIX_MAG, BMIX_MAG
SCF計算における前の電子密度と新しい電子密度との混合(mixing)の方法や度合いを設定する変数. なかなか収束しない場合でも,この変数を変更することで収束にもっていけることもある.
IMIXでmixingの方法を変更できる.
たとえば,
IMIX = 4
AMIX = 0.4
では収束しなかったものでも,
IMIX = 1
AMIX = 0.1
BMIX = 0.01
AMIX_MAG = 0.4
BMIX_MAG = 0.01
とすると収束した経験がある.
KPOINTS
KPOINTSファイルにはk点に関する指定を記述する.
以下のサイトに詳しく記載されているので,ちゃんと読んだほうが良い.
以下は注意点:
- 一般に,金属(metal)の場合にはBrillouin zoneのどこにFermi面が存在しているかなどが重要なため,k点を多く取らなければならない.絶縁体(insulator)や半導体(semiconductor)では,100k-points/atomで十分なことが多いが,金属(metal)ではその10倍以上となることもしばしば.遷移金属(transition metals)の場合,d軌道にFermi面が来たりするとそこら辺のDOSが急峻になるため,5000k-points/atom必要だったりすることもあるらしい.
- Monkhorst-Packで k-point mesh を生成する場合,奇数だとGamma点をとり,偶数だとGamma点をとらない.ゆえに,IRBZ (irreversible Brillouin zone)の中の k-point数は数字の大小だけでは決まらない.FCCの場合,8x8x8の方が,9x9x9よりも多くの k-points / IRBZ となることに注意.ゆえに,8x8x8より小さい場合は偶数にすることがおすすめらしい.それ以上は奇数でも問題ない.
Smearing
上のKPOINTSと関連しているが,電子の占有を決定するためのぼかし方の指定.
- Tetrahedron method with Blochl corrections (
ISMEAR = -5):絶縁体,半導体や,DOSや高精度なtotal energy計算が要求される場合に用いる. - Methfessel-Paxton with N=1 (
ISMEAR = 1) or N=2 (ISMEAR = 2):金属の場合,Gaussian smearingよりも少ないk-pointsと高いぼかしエネルギーで収束.Smearing energy (temperature)は出来る限り大きくすべきだが,free energy と entropy の差が 1-2 meV/atom 程度となるくらいに小さくすべきらしい.
OUTCAR
Energies
下記のような出力の箇所が,SCF iteration毎に出力される. そのうちの最後のものがその計算におけるエネルギーということになる.
----------------------------------------- Iteration 1( 17) ---------------------------------------
POTLOK: cpu time 0.0617: real time 0.0676
SETDIJ: cpu time 0.0123: real time 0.0179
EDDAV: cpu time 21.8159: real time 21.8205
DOS: cpu time 0.0123: real time 0.0065
--------------------------------------------
LOOP: cpu time 21.9146: real time 21.9257
eigenvalue-minimisations : 10416
total energy-change (2. order) :-0.1659891E-06 (-0.8943160E-07)
number of electron 224.0000404 magnetization
augmentation part 14.8966407 magnetization
Free energy of the ion-electron system (eV)
---------------------------------------------------
alpha Z PSCENC = 323.65836222
Ewald energy TEWEN = -8175.04019141
-Hartree energ DENC = -5571.80911778
-exchange EXHF = 0.00000000
-V(xc)+E(xc) XCENC = 788.71002869
PAW double counting = 32617.27325189 -32520.12220369
entropy T*S EENTRO = 0.00000000
eigenvalues EBANDS = -1394.92998503
atomic energy EATOM = 13596.49667107
Solvation Ediel_sol = 0.00000000
---------------------------------------------------
free energy TOTEN = -335.76318405 eV
energy without entropy = -335.76318405 energy(sigma->0) = -335.76318405
--------------------------------------------------------------------------------------------------------
Postprocess
Charge density visualization using VESTA
VESTAという可視化プログラムを利用する. CHGCARとOUTCARを同一フォルダに入れておいて,CHGCARファイルをVESTAへドラッグアンドドロップすれば,表示してくれる(MacOSX).
VASPのCHGやCHGCAR,PARCHGなどのデータは単位なし(電子の個数)となっている(つまり計算セルの体積が掛けられている).VESTAではそれを計算セルの体積で割った値を採用している.その際の体積の単位がBohr^3 なので,VESTAでの電子密度データの値は1/Bohr^3 なので注意.
partial (band decomposed) charge density
すでに計算が行われて収束していること(WAVECARファイルが存在していること)を前提としている.
参考:http://cms.mpi.univie.ac.at/vasp/guide/node145.html
INCARファイルに以下のような行を追加して再計算する.
LPARD = .TRUE.
NBMOD = -2
EINT = 5.67 5.8700616
LSEPB = FALSE
LSEPK = FALSE
LPARD:partial charge densityを計算するというflag.これを立てるとpartial charge densityのみ計算して終了する.NBMOD:どこのpartial charge densityを計算するかという値.- 「-1」はIBANDで指定されたバンドのpartial charge density
- 「-2」はEINTで指定されたenergy range内のpartial charge density
Density of states (DOS)
まず,MDやrelaxationなどを行った結果表示されるようなDOSは「usually useless」らしい.
ISTART = 1
NSW = 0
として再計算したDOSCARを用いるべしとのこと. 実際に高精度なDOSを描きたい場合には,KPOINTSを多少増やさないといけない.SCF計算の収束値(EDIFF)もrelaxation計算よりは厳しくするべきと思われる. その際,
ICHARG = 11
のように,ICHARGに「+10」することでself-consistent計算をしないように設定できる.(これはすべてのelectronic minimizationの間,charge densityひいてはpotentialを固定しておくということ.)
ISMEAR= 0
SIGMA = 0.1 # broadening in eV
NEDOS = 2000 # number of grid points in DOS
EMIN = -20 # minimum energy for evaluation of DOS
EMAX = 15 # maximum energy for evaluation of DOS
ISMEARとSIGMAでsmearingの設定を行う.
ISMEARとして,0(Gaussian) や -1(Fermi分布関数)を指定すると,DOS形状が滑らかになって視覚的には見やすいが,
-5だとより高精度なものとなる.
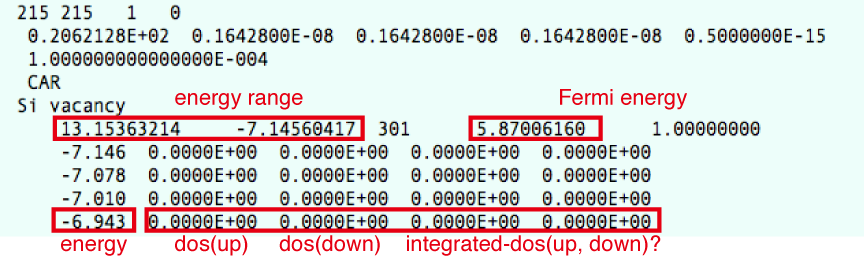
DOSCARというファイルに結果が出力される.spin-polarizedの場合とnon-polarizedの場合で出力が多少異なる.polarizedの場合は,upとdownのDOSが含まれる. spin non-polarized (ISPIN=1)の場合のDOSのリストのところは以下のような記述になっている.
energy DOS integrated-DOS
spin-polalized (ISPIN=2)の場合は,
energy DOS(up) DOS(down) integrated-DOS(up) integrated-DOS(down)
LORBITの値をPDOSを出力するように設定してある場合(LORBIT > 10)は,この後に,
energy s-DOS(up) s-DOS(down) p-DOS(up) p-DOS(down) d-DOS(up) d-DOS(down)
のように出力される(spin-polarizedの場合).
実際のDOSCARファイルの中身はこんな感じ.
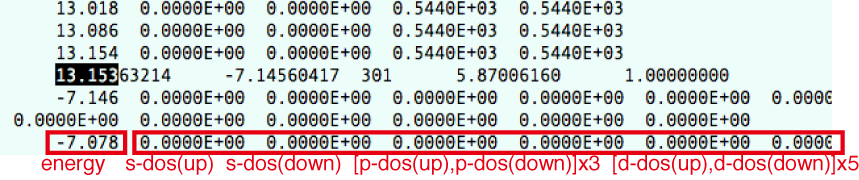
Partial/projected DOS (pDOS)
例えばs軌道やp軌道だけのDOSを描くことができる.
INCARファイルに,
LORBIT = 11
としてDOS計算すると,DOSCARファイルにtotal DOSの下にpDOSが出力される.
ISPIN=2で計算した(スピン分極ありの)場合,spin upのpDOSの下にspin downのpDOSが出力される.
Extract DOS
pymatgenを使ってDOSを抽出するスクリプト.
import numpy as np
from pymatgen.io.vaspio.vasp_output import Vasprun
from pymatgen.electronic_structure.core import Spin
vasprun= Vasprun("./vasprun.xml")
spd_dos= vasprun.complete_dos.get_spd_dos()
pdos= spd_dos['P'].densities[Spin.up]
sdos= spd_dos['S'].densities[Spin.up]
tdos= vasprun.complete_dos.densities[Spin.up]
energies= vasprun.tdos.energies
efermi= vasprun.efermi
f=open('out.dos','w')
for i in range(len(energies)):
f.write(' {0:12.7f} {1:12.7f} {2:12.7f} {3:12.7f}\n'.format(energies[i]-efermi,tdos[i],sdos[i],pdos[i]))
f.close()これを実行すると,out.dosというファイルにDOS情報が出力される.
原子毎のDOSなどを出力したい場合もpymatgenを利用できるかは知らない.
Band structure
バンド図の計算用にいくつかファイルを変更するので, 新しいディレクトリで計算することをお薦めする.
$ mkdir band
$ cp INCAR POSCAR POTCAR KPOINTS CHGCAR band/KPOINTSファイルを以下のように変更する.
band L-G-X <---- comment line
20 <---- num of divisions per segment
line mode <---- tell vasp to use k-points along line
reciprocal
0.5 0.5 0.5 <---- L point
0 0 0 <---- Gamma point
0 0 0 <---- Gamma point
0.5 0.5 0 <---- X point
また,line modeの計算でtetrahedronメッシュが使えないため,INCARファイル内のISMEAR変数を1に設定する(-4や-5以外にする?).
ISMEAR = 1 ! -5 (not be able to use tetrahedron)
あとは普通にvaspを実行する.
結果はEIGENVALに出力される.
ただし,各k点毎に固有値が表示されているので,グラフにするのは面倒.
そこはp4vaspを利用することをお薦めする.
Molecular dynamics (MD)
温度の設定
MDを行う場合,以下のようにパラメータを設定する.
EDIFF = 1.0e-04
ALGO = Very Fast : RMM-DIIS for electrons
IBRION = 0 : MDを指定.
NSW = 10000 : MD step数.
MDALGO = 2 : Nose-Hoover thermostatを指定.
TEBEG = 2000 : 初期温度(K).
TEEND = 2000 : 最終温度(K).
SMASS = 0.4 : Nose-Hoover thermostatのパラメータ.0.4程度?
POTIM = 1.5 : MD time step (fs).
NWRITE = 0 : あまり多くのことを書き出さない
LCHARGE = .FALSE. : 電荷や
LWAVE = .FALSE. : 波動関数は書き出さない
NBLOCK = 10 : XDATCARに10step毎に座標を出力する.
注)
構造最適化などの後の構造からスタートしたい時などに,CONTCARをPOSCARにコピーして使うことがあるが,
構造最適化後には速度が全て0となっており,それが初期値として入力されてしまいTEBEGが有効とならないことがある.
ダイナミクスの可視化
MDした結果,XDATCAR や vasprun.xml に原子位置の情報が書き出されるので,それを抽出してなんらかの可視化ソフトで見れば良い.
ここでは nappy を利用する方法を記載しておく.nappyがインストールされている前提とする.
VASP計算を行ったディレクトリにて,次のように vasprun2fp.py を --sequence をつけて実行すると,vasprun.xmlに含まれているすべてのステップにおける座標情報が 00010/ というような(MDステップ番号の)ディレクトリに出力される.
$ python /path/to/nap/nappy/vasp/vasprun2fp.py --sequencepmdフォーマットとPOSCARフォーマットの2つと他にもいくつかのファイルが出力される.
例えば各ディレクトリのPOSCARファイルを連番ファイルとしてコピーしてくるには,次のようにすればよく,そのディレクトリ内に POSCAR_00010 のような名前のファイルがたくさんできる.
$ for d in 0*; do cp $d/POSCAR POSCAR_$d ; donePSTRESSに関する注意点
ISIF=3と設定し,ionとcell を動くようにしてNpT-MDを行う際に,PSTRESSで外部圧力を設定することができる(PSTRESS in VASP wiki).この時出力されるエネルギーの値には注意が必要.
例えば,とある系に50GPaの外圧をかけてNpT-MDを行った途中のステップの応力部分を次に示す.
FORCE on cell =-STRESS in cart. coord. units (eV):
Direction XX YY ZZ XY YZ ZX
--------------------------------------------------------------------------------------
Alpha Z 236.06822 236.06822 236.06822
Ewald -1560.40175 -1609.33986 -1615.46557 -0.28963 3.40414 23.37784
Hartree 519.92044 525.57855 528.86167 -10.73802 -2.49984 -6.17822
E(xc) -448.72120 -448.22637 -448.23118 0.18285 -0.14943 -0.05898
Local -422.12108 -388.30781 -386.65828 15.30591 -0.47270 -13.95005
n-local -151.58071 -156.21935 -156.02480 -1.09364 1.59950 2.05134
augment 63.73269 64.27313 64.44536 -0.20313 -0.05016 -0.52927
Kinetic 1783.79911 1789.83544 1796.67620 -6.28135 0.86276 -8.86869
Fock 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000
-------------------------------------------------------------------------------------
Total 20.69572 13.66195 19.67162 -3.11701 2.69427 -4.15603
in kB 203.04002 134.03364 192.99282 -30.58010 26.43274 -40.77363
external pressure = -323.31 kB Pullay stress = 500.00 kB
kinetic pressure (ideal gas correction) = 19.53 kB
total pressure = 196.22 kB
Total+kin. 217.465 145.117 226.068 -31.409 26.188 -51.281
Pullay stressというところに外部圧(500 kBar)が書かれていて,external stress = -323.31 kB となっている.これは上で計算された応力のXX, YY, ZZ 項の平均値からPullay stress を引いた値となっている.また,total pressure は,Pullay stressを差し引かずに kinetic pressure を加えた値となっている.
一方,同じ構造を外部圧力を指定せずに単に(MDではなく)SCF計算を行うと,Pullay stressは 0 となり,external pressure は 176.69 となる.
また,NpT-MDのOUTCARには,次のような出力があり,enthalpy HからPV項を差し引いた内部エネルギーの値が明示されている(下では -84.17054764).
FREE ENERGIE OF THE ION-ELECTRON SYSTEM (eV)
---------------------------------------------------
free energy TOTEN = -135.13518603 eV
energy without entropy= -135.13518603 energy(sigma->0) = -135.13518603
enthalpy is TOTEN = -84.17054764 eV P V= 50.96463839
...
...
ENERGY OF THE ELECTRON-ION-THERMOSTAT SYSTEM (eV)
---------------------------------------------------
% ion-electron TOTEN = -84.170548 see above
しかし,PSTRESS を設定しないSCF計算には,上記の free energy TOTENの値しか出力されない.PSTRESS を設定するとそこからPV項として差し引かれたエネルギーが上記のように表示されるようになる.注意すべきなのは,系の圧力(total pressureやexternal pressure)ではなく,Pullay stress値で計算されたPV項が差し引かれている.
これは vasprun.xml 内にも影響している.vasprun.xml内の最後に書かれている e_fr_energy や e_wo_entrp はPV項が差し引かれた値となっている.(scstepエントリー内のe_fr_energyなどはPV項が差し引かれていない.)
Atomic energy
下記サイト参照:
真空に置いた原子1個のエネルギーを計算する場合,以下の様なことを考える.
- 周期的に存在する原子同士が十分離れるように,真空を大きく用意する.(ラフには,2原子分子距離の4〜5倍)
- KPOINTSは一点(ガンマ点)で構わない.(バンド構造を形成しないから.)
- Gaussian smearingを用いる.(テトラヘドロン法はガンマ点のみの計算では使えない.)
energy without entropyを参照する.(状態の縮退があるため,エントロピー項が大きいが考慮しない.)
そのため,KPOINTSファイルは,以下のようになる.
Monkhorst Pack
0
Monkhorst Pack
1 1 1
0 0 0
POSCARファイルは,以下.
atom
1
10.00000 .00000 .00000
.00000 10.00000 .00000
.00000 .00000 10.00000
1
cart
0 0 0
INCARファイルは,以下のようになる.
SYSTEM = Si: atom
ENCUT = 200.00 eV # energy cut-off for the calculation
PREC = Normal # Normal precision
LREAL = .FALSE ! real space projection .FALSE. or Auto
ISMEAR = 0; SIGMA=0.1 use smearing method
ここで,ISMEAR = 0でGaussian smearingを指定している.
NEB (nudged elastic band)計算
NEB計算とは,反応経路の鞍点構造の探索やそのバリアエネルギーの計算を行うもの.Henkelman Groupを参照. 反応や原子拡散の始点と終点をいくつかのイメージで繋いで,それらは仮想的なバネが張られていると考える. バネでつながれた複数のイメージにおいて,始点と終点を固定して,全体の安定構造を求めようとすると,中間のバネとイメージが反応経路の鞍点の近くを通るものとなる. 固体中の原子拡散の反応経路探索には非常に良く使われる.
VASP(5.4.1)でNEB計算を行うには,以下の準備が必要.
- 始点と終点の構造.これらはNEB計算の間は固定点として扱われるので,それぞれで構造最適化を行う必要がある(そうしないと,安定点と鞍点とのエネルギー差を正確に見積もることができない).
- 始点と終点の間の構造を作成する.この中間構造はあくまでNEBの初期構造なので,簡単に始点と終点の対応する原子の座標の補間(interpolation)でかまわない.対称的な経路の場合には,全部で奇数個のイメージを用意しないと中心のイメージが鞍点とならない.また,周期境界条件の場合,始点と終点において対応する原子が周期境界をまたいでしまうと,原子の移動がシミュレーションセルサイズと同程度となってしまうことがあるので注意が必要.
- VASP計算用のディレクトリを用意する.
00から09のような名前のディレクトリをイメージの数(始点と終点を含む)だけ用意する.つまりこの場合,00には始点,09には終点の構造を置くことになる. INCARファイルを作成する.以下にサンプルを載せる.ISTART = 1 ICHARG = 1 INIWAV = 1 ISPIN = 1 ISYM = 0 !<=== おそらく対称性の制限を外した方が良い ENCUT = 400.000 LREAL = Auto EDIFF = 1.0e-04 ALGO = Fast PREC = Normal NELMIN = 4 NELM = 100 NBANDS = 98 ISMEAR = 2 SIGMA = 0.2 IBRION = 1 !<=== RMM-DIIS (1) or quick-min (3) が推奨されている NSW = 100 ISIF = 2 NCORE = 8 # NEB OPTIONS IMAGES = 7 !<=== 始点と終点を除くイメージ数 SPRING = -5.0 !<=== バネ定数 LCLIMB = .TRUE. !<=== climbing image NEBを行うかどうか- VASP計算をMPIで実行する際,実際に計算する(始点と終点を除く)イメージ数 x 各イメージの並列数の数のMPI並列数を指定して実行する.