🎥 使い方動画(YouTube)
サンプルの実行
example/test-W/ ディレクトリにはいくつかの入力ファイル(in.pmd および pmdini)がある.これらの入力ファイルは,体心立方(BCC)タングステン結晶構造に 1 つのヘリウム原子を含むシステムのもの.
次のようにして pmd コマンドを実行すると 100 ステップの NVE-MD シミュレーションが実行される.
cd example/test-W
../../pmd/pmd総エネルギー,運動エネルギー,ポテンシャルエネルギーは out.erg ファイルに出力される.
これらのエネルギーの変化を gnuplot を使用して可視化できる.
$ gnuplot
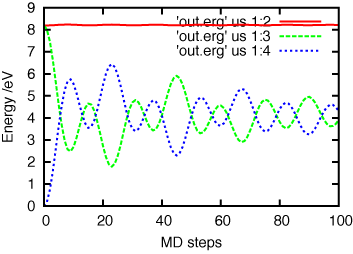
gnuplot> plot 'out.erg' us 1:3 w l, 'out.erg' us 1:4 w l, 'out.erg' us 1:5 w lこの場合,NVE-MD シミュレーションを実行しているため,総エネルギーは運動エネルギーとポテンシャルエネルギーの和となり保存される.
NOTE
out.ergでは総エネルギーとポテンシャルエネルギーは初期値を差し引かない生の値になっている.
また,100 ステップのうち 10 ステップごとに原子構造が LAMMPS-dump 形式で出力される(例:dump_0,dump_10,dump_100 など).
pmd の実行に必要な入力ファイル
pmd を実行するには,作業ディレクトリ内に以下のファイルが必要.
in.pmd:シミュレーション設定を記述した入力ファイル(詳細は04_in-pmd)pmdini:セル情報,原子の初期位置および速度を記述したファイル(詳細は03_pmd-file)
また,一部の原子間ポテンシャルを使用する場合は,in.params.xxx のようなポテンシャル固有のパラメータファイルも必要になる(Coulombポテンシャルの場合に in.params.Coulomb となる).
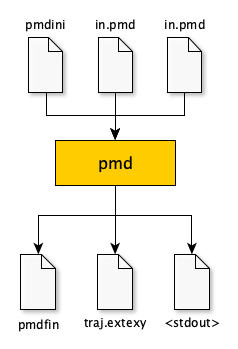 pmd が実行されると,次のファイルが出力される.
pmd が実行されると,次のファイルが出力される.
pmdfin:最終構造(pmdフォーマット)traj.extxyz:途中の構造(フォーマットや出力頻度は in.pmd で指定する)(v0.13.0以降)- 上図の
<stdout>は標準出力(画面出力)なので,リダイレクトしてout.pmdなどに出力する.$ pmd | tee out.pmd または $ pmd > out.pmd
pmd で使用される単位
- 長さ:オングストローム(Å)
- 時間:フェムト秒(fs)
- エネルギー:電子ボルト(eV)
- 質量:炭素原子の 1/12
初期原子構造ファイルの作成
詳細は 03_pmd-file を参照のこと.
pmd を実行するには,初期原子構造ファイル pmdini を用意する必要がある.
すでにいくつかの構造を作成するプログラム(nappy/mkcell/cell_maker.py)がある.
それらのコードを参考にして独自の構造ファイルを作成することもできる.
上記の中に目的のシステムの原子構造ファイルを作成するプログラムがある場合は,以下のように実行する.
$ cd mkconf
$ emacs makefile
# 作成する mkconf_* の確認
$ make mkconf_Si_disl
$ ./mkconf_Si_dislまたは,nappy/mkcell/cell_maker.py を使用することもできる.
$ python /path/to/nappy/mkcell/cell_maker.py -h
...
$ python /path/to/nappy/mkcell/cell_maker.py dia -l 5.427 -s 4,4,4これにより pmdini ファイルが作成される.
NOTE
原子構造ファイルを作成するプログラムを新規に作成する場合,
mkconf_BCC.Fなどのプログラムをコピーして修正し,makefileにエントリを追加してコンパイルする.
in.pmd ファイルの作成
in.pmd ファイルの詳細については 04_in-pmd を参照する.
例えば,SW_Si ポテンシャルを使用した 1000 ステップの MD シミュレーションの in.pmd ファイルの例は以下のとおり.
io_format ascii
print_level 1
time_interval 2d0
num_iteration 1000
num_out_energy 100
num_out_pmd 10
force_type SW_Si
cutoff_radius 3.7712d0
cutoff_buffer 0.2d0
! や # で始まる行はコメントとして扱われ,空白行は無視される.
pmd の実行
1 プロセスでの実行
in.pmd と pmdini があるディレクトリで,以下のコマンドを実行すると pmd を実行できる.
$ /path/to/pmd/pmd | tee out.pmdバックグラウンドで実行する場合は以下のようにする.
$ /path/to/pmd/pmd > out.pmd 2>&1 &pmd を実行すると,以下のファイルが出力される.
out.erg:総エネルギー,運動エネルギー,ポテンシャルエネルギー,温度,体積,圧力traj.extxyz:特定の MD ステップの原子構造
MPIを用いた複数プロセス(ノード)実行
pmdはMPIコンパイラを使ってコンパイルされているので,MPIを使った並列実行が可能.次のように mpirun(もしくは mpiexec)コマンドを使って並列実行する.
$ mpirun -np 4 /path/to/pmd/pmd | tee out.pmdシステムサイズが小さすぎると領域分割できないためにMPI並列実行ができない場合がある.
OpenMPを使ったプロセス並列実行
OpenMPを有効にしてコンパイルした場合は,次のようにして OpenMP を使ったプロセス並列実行が可能.ただし,ノードをまたいだ並列実行はできない.また,あまりに多い並列数は並列化効率が下がるので,並列数を増やすのであればMPI並列の方が有意.
$ OMP_NUM_THREADS=2 mpirun -np 2 /path/to/pmd/pmd | tee out.pmd