Run pmd¶
Run a sample simulation¶
There are some input files in example/test-W/ directory. ( in.pmd
and pmdini ) These input files are for the system of BCC tungsten
crystalline structure including one helium atom.
$ cd example/test-W
$ ../../pmd/pmd
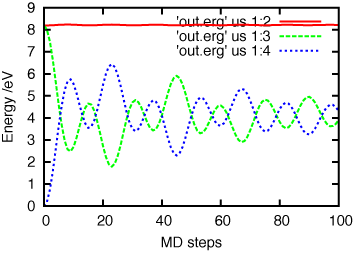
When you run the pmd command like above, NVE -MD simulation of 100
steps is performed. And the total, kinetic, and potential energies are
output in out.erg file. So you can look at the evoluation of these
energies using gnuplot command as,
$ gnuplot
gnuplot> plot 'out.erg' us 1:3 w l, 'out.erg' us 1:4 w l, 'out.erg' us 1:5 w l
In this case, since you are performing NVE -MD simulation of bcc-W, the total energy conserves conpensating the deviations of kinetic and potential energies.
Note
The format ot out.erg is a bit changed from that of before 2018-11-01
versions. The total and potential energies are raw values not being
subtracted the initial values.
And also configurations of atoms at each 10 steps out of 100 steps are
written in LAMMPS-dump format, e.g., dump_0, dump_10,...,
dump_100.
Input files needed to run pmd¶
To run pmd, the following files are required in the working directory,
in.pmd-- Input file that describes simulation setting.pmdini-- Cell information and initial positions and velocities of atoms.
And there are some optional files required by the pmd if you use
interatomic potentials that require input parameters from files such as
in.params.xxx.
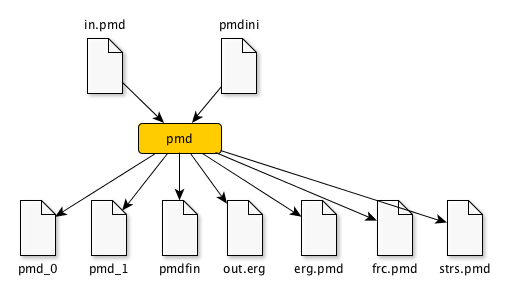
After running pmd , some output files appear in the same directory.
Units used in pmd¶
- Length: Angstrom
- Time: femto second (fs)
- Energy: electron volt (eV)
- Mass: 1/12 of carbon atom
Make an initial atom-configuration file¶
Please Atom-configuration file for detail.
One has to make an initial atom-configuration file, pmdini, to run
pmd. There are already some programs that make initial
atom-configuration files of some systems (mkconf/mkconf_????.F and/or
nappy/mkcell/cell_maker.py). You can make your own initial
atom-configuration file by looking at those program codes.
If there is already a program that makes an atom-configuration file of your target system, you can make an atom-configuration file as,
$ cd mkconf
$ emacs makefile
(find which mkconf_* will be made)
$ make mkconf_Si_disl
$ ./mkconf_Si_disl
or you can use nappy/mkcell/cell_maker.py as well,
$ python /path/to/nappy/mkcell/cell_maker.py -h
...
$ python /path/to/nappy/mkcell/cell_maker.py dia -l 5.427 -s 4,4,4
Then you get an atom-configuration file pmdini.
Note
If you have to make the program which makes an atom-configuration file,
copy any program like mkconf_BCC.F , modify it, add an entry into
makefile , and compile.
Make the in.pmd file¶
Please refer in-pmd{.interpreted-text role="ref"} for details of
in.pmd file.
For instance, in.pmd file for the system of 1000 step MD simulation
using SW_Si potential is as follows,
#
# unit of time = femto sec
# unit of length= Angstrom
# unit of mass = unified atomic mass unit
#
io_format ascii
print_level 1
time_interval 2d0
num_iteration 1000
num_out_energy 100
flag_out_pmd 1
num_out_pmd 10
force_type SW_Si
cutoff_radius 3.7712d0
cutoff_buffer 0.2d0
flag_damping 2
damping_coeff 0.5d0
converge_eps 1d-4
converge_num 3
initial_temperature -2000d0
final_temperature -2000d0
temperature_control none
temperature_target 100d0
temperature_relax_time 1d0
factor_direction 3 2
1.000d0 1.000d0 1.000d0
1.000d0 0.000d0 1.000d0
stress_control none
stress_relax_time 100d0
stress_target
0.00d0 0.00d0 0.00d0
0.00d0 0.00d0 0.00d0
0.00d0 0.00d0 0.00d0
pressure_target 1.00
shear_stress 0.00
Here, the lines begin with ! or # are treated as comment lines and
blanc lines are skipped.
Run pmd¶
Run pmd on 1-process¶
It is really easy to run pmd on 1-process. On the directory where
in.pmd and pmdini exist, just execute pmd as,
$ /path/to/pmd/pmd
If you want to perform it background,
$ /path/to/pmd/pmd > out.pmd 2>&1 &
The following files appear when you perform pmd,
out.erg-- Total, kinetic, potential energies, and temperature, volume, pressure.dump_##-- Atom-configurations at a certain MD step is written in LAMMPS-dump format by default.##means the MD step.
Run pmd on parallel-nodes (MPI and OpenMP)¶
Different from the old version of pmd which requires divided atom configuration files for parallel nodes, in the current version (since 2016-05-05), the parallel simulation can be performed almost the same as the serial run.
Just you need to describe how many divisions on each direction in
in.pmd such as num_nodes_x, num_nodes_y and num_nodes_z ,and run
pmd with mpirun or mpiexec command to run MPI executable.
$ mpirun -np 8 /path/to/pmd > out.pmd 2>&1 &
Here, pmd will be executed on 8-nodes and the standard output is
written into out.pmd.
If you want to run the pmd using OpenMP, assuming that the pmd is compiled with OpenMP option, you can run it as,
$ OMP_NUM_THREADS=4 /path/to/pmd > out.pmd 2>&1 &
or you can also combine OpenMP with MPI as,
$ export OMP_NUM_THREADS=8
$ mpirun -np /path/to/pmd > out.pmd &
Currently (2022/04/22), only several selected force-fields are implemented with OpenMP, such as Morse, Coulomb, angular, and RFMEAM.
If any job-scheduling system is available on the system you are using, describe the above command in your job script to be submitted.
Tip for large-scale simulation¶
When performing large-scale MD simulations over 10,000 atoms, the size of files that contain atom information gets considerably large and the cost for transfering those files from a remote server to the local machine increases. One can reduce the file size using gzip command in Linux/Unix/Mac as,
$ gzip dump_0
which changes the text file, dump_0, to a binary file, dump_0.gz.
The python utility in nap, nappy, and the visualization software, ovito, can read gzipped files in the same manner as text files.